Vectors, Plasmids and the CGT Trap: APAC's Cell and Gene Therapy Ambitions Face an Upstream Bottleneck
02 July 2026 | Thursday | Analysis

THE GATE
The choke point nobody puts on the slide
Somewhere in the region this quarter, a cell-and-gene developer has everything the pitch deck promised. The Phase I data is clean. Patients are enrolled. A regulator — the HSA, the CDSCO, the PMDA — has shown interest. And the programme is stalled anyway, not in the clinic but in a queue: a wait for a slot on a GMP viral-vector line. The therapy exists on paper. The vector does not yet exist in a vial with a certificate of analysis. Until it does, there is no therapy — only a plan for one.
That gap between an approved-looking programme and an actual vial is the story of APAC’s cell-and-gene ambition. Across the region the binding constraint on that ambition is not clinical demand, not scientific talent, not even capital. It sits upstream, in the supply of viral vectors — adeno-associated virus (AAV), lentivirus, adenovirus — and the plasmid DNA that every one of them is built from. Vectors are the delivery vehicle for roughly seven in ten cell and gene therapies. Secure the vector and you have a medicine. Miss the slot and you have a slide.
There is a paradox to reconcile first, because it confuses newcomers. At the global level you hear two contradictory stories. One says viral-vector capacity is in oversupply — CDMO suites built during the pandemic boom now running below half utilisation, because clinical demand is lumpy and there are not yet enough commercial products to keep 2,000-litre bioreactors busy. The other says there is a chronic shortage, with developers waiting the better part of a year for a slot. Both are true, and the way they fit together is exactly what matters for APAC.
The overcapacity is concentrated in early-stage, clinical-grade work, largely in the United States and Europe, and — the crucial word — outside the region. The scarcity is in GMP-grade, commercial-ready, comparability-stable, and region-local supply. For a developer in Singapore, Seoul, Mumbai or Melbourne, spare capacity in Pennsylvania or on the Genopole campus is not the same as capacity you can reach, afford, schedule around and hold a stable process against. Vector you cannot get to on your timeline is not, in any operational sense, available vector.
Meanwhile the ambition is real and it is arriving nearly everywhere at once. On the strength of advanced-therapy trial activity, Asia-Pacific has become the second-largest region for such trials, ahead of Europe. Singapore, South Korea, Japan and Australia have each engineered deliberate ecosystems; India has moved in a single decade from spectator to producer. The cell-and-gene market itself is accelerating — by one industry count from about US$21.3 billion in 2025 to US$26.2 billion in 2026 — with APAC the fastest-growing slice of it. Yet pipeline depth is wildly uneven. India has logged fewer than ten cell-and-gene-focused clinical trials against more than 150 in the United States and more than 160 in China. The clinical ambition, in other words, is running well ahead of the upstream that has to feed it.
Secure the vector and you have a medicine. Miss the slot and you have a slide.
Because the vector is the gate, the decision about how to get it — build your own capability, buy a slot from a contract manufacturer, or push production out to the point of care — quietly decides whether a programme is viable at all. It looks like a supply question. It is really a manufacturing-strategy question wearing a supply question’s clothes, and it is being answered, market by market, in four very different accents.
THE REALITY
Capacity, cost and the comparability trap
Start with the number almost no one puts on a slide: the wait. A qualified GMP vector slot in the region is routinely quoted at close to a year — which, tellingly, is roughly the same time it now takes to build a modular vector suite from scratch (ten to twelve months). When your slot is a year out and your clinical window for a fast-tracked orphan indication is measured in months, the queue is the programme risk. Everything else is secondary to it.
Then the cost. Single viral-vector batches can exceed US$500,000, a figure aired at the 2026 cell-and-gene congress in Seoul. Vectors are structurally more expensive to make than monoclonal antibodies: the molecules are larger and more complex; they demand BSL-2 containment where a typical antibody needs only BSL-1; starting titres and overall process recoveries remain low, as they were for antibodies in their early years; and biosafety pushes everything toward single-use consumables. That cost cascades all the way to the patient. A centralised autologous CAR-T can run US$300,000 to US$500,000 per patient, and some individual gene therapies are priced as high as US$3.5 million. For an APAC market that has to reach patients at a fraction of Western prices, the vector line is precisely where the economics are won or lost.
Batch-failure risk is the third pressure, and the cruellest, because autologous therapies are single-patient batches. There is no lot to fall back on. A failed vector lot or a failed transduction is not a yield statistic on a dashboard; it is a patient — often a heavily pretreated patient who cannot wait for a re-run — left without a therapy. Lentiviral vectors are notoriously fragile, sensitive to shear, pH, salt concentration, temperature and buffer osmolarity as they move through clarification, chromatography, and ultrafiltration and diafiltration. Every fragile step upstream is a failure mode waiting downstream.
And then the beat that separates people who have shipped product from people who have drawn org charts: comparability. Switch your vector source — from an academic vector core to a CDMO, from CDMO A to CDMO B, from an adherent process to suspension, from one plasmid supplier to another — and you inherit a comparability exercise. Analytical comparability across identity, purity, potency, vector-genome titre, infectious titre and, for AAV, the empty-to-full capsid ratio, must be re-established, and regulators will ask you to prove it. A vector “shortage” is therefore not only about volume. It is about the switching cost of chasing volume.
That is the trap in the headline. Developers get locked into a supplier not by contract but by comparability, which quietly converts a capacity problem into a strategic-dependency problem. The moment your process is validated against one source’s vector, moving becomes a data package, a regulatory conversation and months of your life — exactly what a developer racing a clinical window does not have. Capacity you cannot switch to without a comparability bridge is capacity you do not really have either.
Underneath all of it sits a talent-and-analytics scarcity that no bioreactor purchase fixes. The regional gap is not only steel and single-use plastic; it is vector biology, process analytics and the QC capacity to release product. In India, practitioners flag vector biology specifically as the scarcest expertise in the field. Region-wide, standardised methods and raw-material traceability remain immature — and that immaturity is exactly what prolongs timelines and inflates cost for the early-stage developers who can least absorb either. As one process-development lead at a Singapore vector CDMO put it to this series, “we are not short of ambition or even of capital; we are short of people who have made a hundred lentivirus batches and know why the ninety-ninth failed.”
THE DECISION
Make, buy, or vertically integrate
This is the classic build-versus-buy of biomanufacturing, but harder, because procuring vector capacity carries more complexity and more risk than a routine outsource — the comparability lock-in and the biosafety-facility burden see to that. Three postures are now visible across APAC, and each has become a kind of national signature.
Buy — the CDMO route. South Korea is the region’s clearest bet on scale-through-CDMO. SK pharmteco has built an AAV, lentivirus and adenovirus platform business and, importantly, a dedicated plasmid suite — plasmid being the upstream-of-the-upstream raw material — positioning one of Korea’s largest conglomerates as a vector supplier to the world. But the tell is in the geography: much of SK’s marquee vector capacity physically sits in France (through Yposkesi) and the United States (through the Center for Breakthrough Medicines), with the King of Prussia plasmid suite added stateside and a French commercial vector plant CGMP-qualified only in 2026. It is a Korean champion whose GMP vector footprint remains substantially ex-region — which is the “region-local” nuance made concrete.
The counter-signal is just as instructive. Samsung Biologics, the region’s antibody-CDMO giant, has repeatedly declined to stake its expansion on cell-and-gene or multimodal capacity, judging the market not yet mature enough for large-scale production and profit and choosing to build more antibody plants instead. When the region’s most capable manufacturer keeps its money on antibodies, it is saying something about the commercial pull behind vectors today. Singapore, for its part, plays the specialist-hub role: vector CDMOs such as CellVec (HSA-certified, PIC/S-aligned, running lentivirus and AAV), A*STAR’s Bioprocessing Technology Institute developing immune-cell and vector processes, and Cytiva’s Singapore training lab working directly on the talent gate. Small domestic patient base, high quality and harmonisation — a hub, not a volume play.
Control the plasmid and you de-risk the vector. Control the vector and you de-risk the therapy.
Build — the vertical-integration model. India’s answer is to own the whole chain. ImmunoACT, the IIT-Bombay spin-out behind NexCAR19 — actalycabtagene autoleucel, India’s first approved CAR-T — runs an integrated line from plasmid design to lentiviral vector to ex-vivo cell engineering, in-house. The result is a production cost of roughly US$15,000 per patient against US$300,000 to US$500,000 for Western equivalents, and a plan to scale from about 750 toward 1,500 patients a year. Backed by the Department of Biotechnology’s BioE3 policy, it is standing up a 200-litre GMP lentiviral-vector and plasmid platform targeting on the order of 1,000 patients annually.
India’s broader plasmid-DNA push — the deliberate decision to make the raw material domestically rather than import it — is the upstream story in miniature. Control the plasmid and you de-risk the vector; control the vector and you de-risk the therapy; and if you do all three, you own your cost of goods rather than renting it from someone else’s queue. Other Indian entrants are circling the same logic from different angles: Immuneel, Dr Reddy’s, Intas, Cipla and Natco on the commercial side, Tata Memorial exploring CAR-NK. The through-line is that affordability in India is not a discount; it is an architecture, and the architecture is integration.
The modular middle path. Between a year-long CDMO queue and a greenfield facility sits the modular, single-use, turnkey vector suite — commissionable in ten to twelve months, keeping process and IP control in-house while reaching commercial readiness. For emerging developers without deep facility-engineering muscle, modular is the pragmatic hedge: faster than building conventionally, more controlled than outsourcing, and increasingly the default for a platform company that expects to run many programmes rather than one.
So when does build win? The calculus turns on a handful of variables: how many programmes you are running (one asset argues for buy, a platform argues for build); how tight your speed-to-a-fast-tracked-window is; what cost-of-goods target the market you serve will bear; and how much comparability risk you are willing to hand to a third party. A developer aiming at an early exit or a large-pharma partnership outsources. A developer building a durable, price-competitive APAC franchise integrates. The chief technology officer of one regional CGT developer framed the decision bluntly for this series: “the day we realised our valuation lived and died on a vendor’s production schedule was the day we decided to build.” The uncomfortable through-line for everyone: in cell-and-gene, manufacturing is not downstream of the therapy. Increasingly, it is the therapy.
THE WORKAROUND
Point of care: decentralising the cell, not the vector
If you cannot get centralised capacity, the intuitive move is to bring manufacturing to the hospital. Automated closed systems — the CliniMACS Prodigy is the archetype — have made in-hospital, point-of-care CAR-T a realistic option for academic centres that could never justify a full commercial suite. The clinical case is genuinely strong. Fresh, non-cryopreserved product tends to expand faster and more robustly in the patient; cold-chain and shipping costs collapse; and vein-to-vein logistics simplify dramatically when the cells never leave the building.
The APAC evidence is accumulating. India’s VELCART trial, run at Christian Medical College Vellore with Miltenyi’s Lentigen, manufactured anti-CD19 CAR-T on-site on the Prodigy for ten relapsed or refractory patients, reporting complete remission in all of the B-ALL cohort and half of the DLBCL cohort, every patient infused with fresh product — a clean proof of decentralised feasibility. Thailand’s Siriraj Cell Factory has run a comparable single-centre pilot with its own SiCF-019 construct. Australia offers the institutional gold standard: Cell Therapies Pty Ltd at the Peter MacCallum Cancer Centre is the country’s only TGA-licensed GMP facility for both commercial and clinical cell therapy, manufactures Novartis CAR-T locally with more than a hundred patients treated, and even holds Japanese MHLW accreditation. Japan’s own route runs partly through contract sites such as the FBRI in Kobe. The pattern is unmistakable: the cell-processing step decentralises well.
Point-of-care relieves the cold-chain bottleneck. It does not, by itself, relieve the vector-and-plasmid bottleneck.
Here is the catch the enthusiasts skip. Point-of-care decentralises the cell-processing step — leukapheresis, transduction, expansion, fill. It does not, on its own, decentralise the vectorology. Those Prodigy runs still consume a lentiviral vector supplied from somewhere; in VELCART, that somewhere was Lentigen. Point-of-care relieves the cold-chain and the facility-slot bottleneck. It does not relieve the vector-and-plasmid bottleneck unless the region also solves upstream supply. Decentralised cell manufacturing without decentralised — or at least secured — vector supply simply relocates the queue one step to the left.
And it strains every regulatory framework in the region, each of which has answered the decentralisation question differently. In Japan, the PMDA and MHLW treat ex-vivo gene-modified cells as “regenerative medical products” under the PMD Act, with a conditional and time-limited approval pathway and a separate Act on the Safety of Regenerative Medicine governing in-hospital processing; the 2024 reversal of HeartSheet — the first conditionally approved product denied full approval after post-marketing data — was a pointed signal that the scheme is enforced, not a rubber stamp. South Korea works through its Advanced Regenerative Medicine and Advanced Biopharmaceuticals framework, in force since 2020. Singapore layers HSA’s cell-tissue-and-gene-therapy product classification over specific Ministry of Health licence conditions for hospitals administering in-house-manufactured product, while co-chairing advanced-therapy harmonisation through the ACCESS Consortium. Australia’s TGA is actively working the hospital-manufacturing question through its personalised- and custom-made-device reforms and a dedicated point-of-care consultation.
The unresolved core is the same comparability trap, multiplied by geography: how do you guarantee product equivalence and site-to-site consistency across many small, decentralised sites when standardised methods still do not exist? An APAC regulator familiar with decentralised-manufacturing policy put the official posture plainly for this series: “we want point-of-care to work, because access depends on it — but the burden of proving equivalence sits with the manufacturer, and right now that burden is heavy, and it should be.”
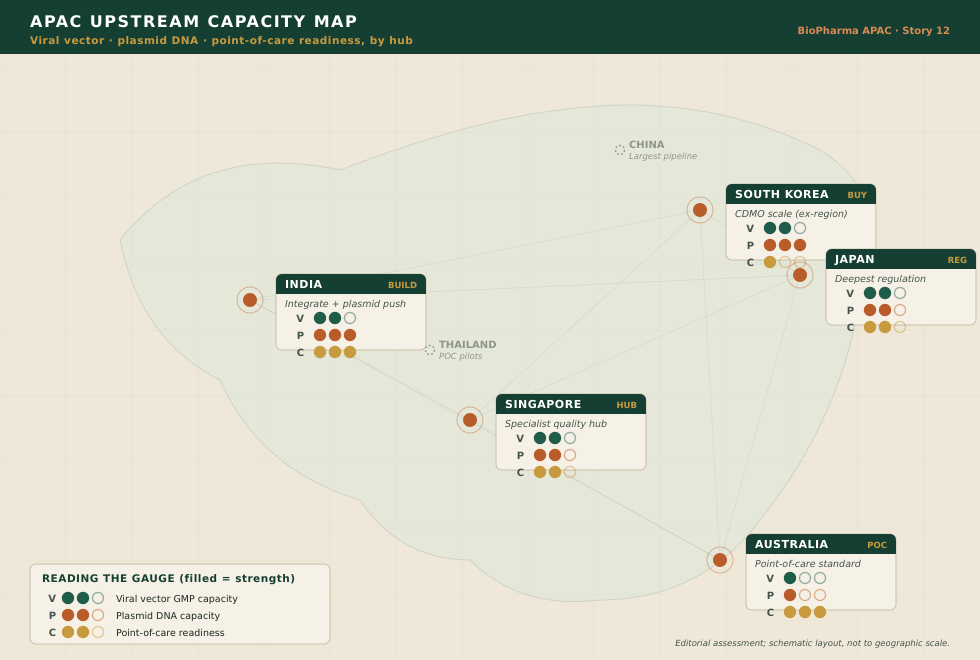
THE MAP
Reading APAC’s upstream capacity
Laid over a map, the region resolves into five distinct upstream strategies rather than one race. The accompanying capacity map plots them; the readout below is the key.
|
MARKET |
UPSTREAM POSTURE |
|
Singapore |
Specialist vector CDMOs plus A*STAR process development and a training pipeline; strong regulatory harmonisation. Small domestic patient base — plays the quality-hub role, not the volume play. |
|
South Korea |
Conglomerate-scale CDMO ambition (SK pharmteco) and plasmid suites — but marquee GMP vector capacity still substantially ex-region. Samsung’s abstention is a caution on commercial pull. |
|
India |
Vertical integration plus a domestic plasmid push; the lowest cost-of-goods globally and a working commercial CAR-T. Thin trial pipeline, but a decisive affordability edge. |
|
Japan |
The deepest regenerative-medicine regulatory architecture and a conditional pathway; contract and hospital manufacturing nodes. Enforcement (HeartSheet) signals maturity, not leniency. |
|
Australia |
The point-of-care / institutional gold standard (Cell Therapies at PeterMac); TGA actively regulating decentralised manufacture. Access-led, quality-anchored. |
|
Regional frame |
China’s large pipeline and Thailand’s point-of-care pilots bracket the region — the demand and the decentralisation extremes between which the four hubs are positioning. |
THE TAKEAWAY
Written upstream
Strip away the market maps and the ribbon-cuttings and the position is simple. APAC’s clinical ambition has outrun its upstream supply. The therapies work; the science travels; the patients are there and waiting. What is scarce is the vector and the plasmid — and, underneath those, the comparability discipline, the vector-biology talent and the region-local GMP capacity to make them repeatable, batch after single-patient batch.
Three strategies are in play. Buy: CDMO scale, most visibly in Korea. Build: vertical integration, most convincingly in India. Decentralise: point-of-care, most maturely in Australia, and increasingly in India and Japan. None of the three removes the upstream gate on its own. Each simply moves it — the CDMO route hands the gate to a vendor’s schedule, integration converts it into a capital-and-talent problem you now own, and point-of-care slides it one step left to the vector still arriving from off-site. The gate does not disappear. It changes hands.
Which is why the region that stops treating vector-and-plasmid capacity as a procurement line item, and starts treating it as core infrastructure — the way it once treated fabs, ports and grid — will set the pace for everyone else. Upstream capacity is not a cost centre attached to a therapy; in cell-and-gene, it is the therapy’s foundation, and it behaves like infrastructure: expensive to build, slow to mature, and decisive once it exists.
Until then, APAC’s cell-and-gene future is written upstream — in vials that either exist, with a certificate of analysis attached, or do not. Whether the region closes that gap or stays gated at the upstream step while the clinic keeps sprinting ahead is the single question that will decide the next decade of APAC advanced therapy. Everything downstream — the trials, the approvals, the patients waiting — is already ready. It is the upstream that has to catch up.
arcilla.fran@biopharmaapac.com
DISCLAIMER
This feature is an editorial and analytical work of journalism produced by BioPharma APAC and is provided for information and industry-commentary purposes only. It does not constitute investment, financial, medical, clinical, legal or regulatory advice, nor a recommendation to buy, sell, prescribe or engage with any product, therapy, company or security named. Market sizes, capacity assessments, lead times, batch costs and cost-per-patient figures are directional, are drawn from publicly available sources and industry commentary believed reliable as of the date of publication, and are subject to change; readers should verify all figures independently before relying on them. References to specific companies, facilities, trials, products and regulatory frameworks are included for illustration and analysis and do not imply any endorsement, affiliation, or independent verification of any organisation’s claims. Except where a source is expressly identified by name, attributed remarks represent composite, representative industry perspectives compiled for this series and should not be read as verbatim statements of, or attributed to, any specific individual or organisation. BioPharma APAC is an independent editorial title and is solely responsible for this content.
© 2026 BioPharma APAC. All rights reserved. No part of this article may be reproduced, distributed or transmitted in any form without prior written permission.
Most Read
- How Does GLP-1 Work?
- Innovations In Magnetic Resonance Imaging Introduced By United Imaging
- Management of Relapsed/Refractory Multiple Myeloma
- 2025 Drug Approvals, Decoded: What Every Biopharma Leader Needs to Know
- BioPharma Manufacturing Resilience: Lessons From Capacity Expansion and Supply Chain Resets from 2025
- APAC Biopharma Review 2025: Innovation, Investment, and Influence on the Global Stage
- Top 25 Biotech Innovations Redefining Health And Planet In 2025
- The New AI Gold Rush: Western Pharma’s Billion-Dollar Bet on Chinese Biotech
- Single-Use Systems Are Rewiring Biopharma Manufacturing
- The State of Biotech and Life Science Jobs in Asia Pacific – 2025
- Asia-Pacific Leads the Charge: Latest Global BioSupplier Technologies of 2025
- Invisible Threats, Visible Risks: How the Nitrosamine Crisis Reshaped Asia’s Pharmaceutical Quality Landscape
Bio Jobs
- Sanofi Turns The Page As Belén Garijo Steps In And Paul Hudson Steps Out
- Global Survey Reveals Nearly 40% of Employees Facing Fertility Challenges Consider Leaving Their Jobs
- BioMed X and AbbVie Begin Global Search for Bold Neuroscience Talent To Decode the Biology of Anhedonia
- Thermo Fisher Expands Bengaluru R&D Centre to Advance Antibody Innovation and Strengthen India’s Life Sciences Ecosystem
- Accord Plasma (Intas Group) Acquires Prothya Biosolutions to Expand Global Plasma Capabilities
- ACG Announces $200 Million Investment to Establish First U.S. Capsule Manufacturing Facility in Atlanta
- AstraZeneca Invests $4.5 Billion to Build Advanced Manufacturing Facility in Virginia, Expanding U.S. Medicine Production
News
Editor Picks



